IgA Vasculitis
IgA Vasculitis
(Henoch-Schönlein purpura)
IgA vasculitis is a systemic disease characterized by inflammation of the small blood vessels. It is formerly known as Henoch-Schönlein purpura, however due to the better reflect the underlying pathophysiology of the disease, it was renamed as IgA vasculitis (IgAV) by the International Chapel Hill Consensus Conference.
Pathophysiology
Hallmark : Deposition of IgA1-domminant immune complexes in the small vessels of the skin, kidneys, joints and GI tract (rarely CNS & lungs), causing inflammation & organ-specific symptoms.
It involves a complex interactions between the immune system, genetic predisposition, & environmental triggers.
Underlying mechanism (The “multi-hit” model) :
- Abnormal IgA1 formation (central features) – Defective O-glycosylation (Aberrant glycosylation) produces galactose-deficient IgA1 (Gd-IgA1)
- Immune complex formation – Abnormal Gd-IgA1 are recognized as autoantigen, triggering the autoantibodies (IgG/IgA) production & formation of immune complexes.
- Deposition & inflammation – These immune complexes then deposit in small vessels of the skin, joints, gut & kidneys causing complement activation (alternative & lectin pathway), inflammatory cells recruitment & eventually tissue damage due to the inflammatory processes.
The sequence of events typically occurs in individuals with a genetic predisposition, primarily linked to specific Human Leukocyte Antigen (HLA) alleles, such as HLA-DRB1 when they are exposed to environmental triggers.
Common triggers include URTI and sometimes GI infection.
Infective triggers (most common) :
- URTI – especially Group A Streptococcal
- Viral infections – infectious mononucleosis, adenovirus, parvovirus, varicella-zoster, rotavirus.
- GI pathogens – Campylobacter, Yersinia, Shigella, Salmonella
- Others – Mycoplasma pneumoniae, Hepatitis, H. pylori, Infective endocarditis, Legionella, brucellosis, etc.
Potential non-infective triggers :
- Drugs, e.g. ampicillin, erythromycin, penicillin, quinidine, quinine, losartan & cytarabine
- Vaccines
- Foods
- Cold temperature
- Insect bites
Clinical features
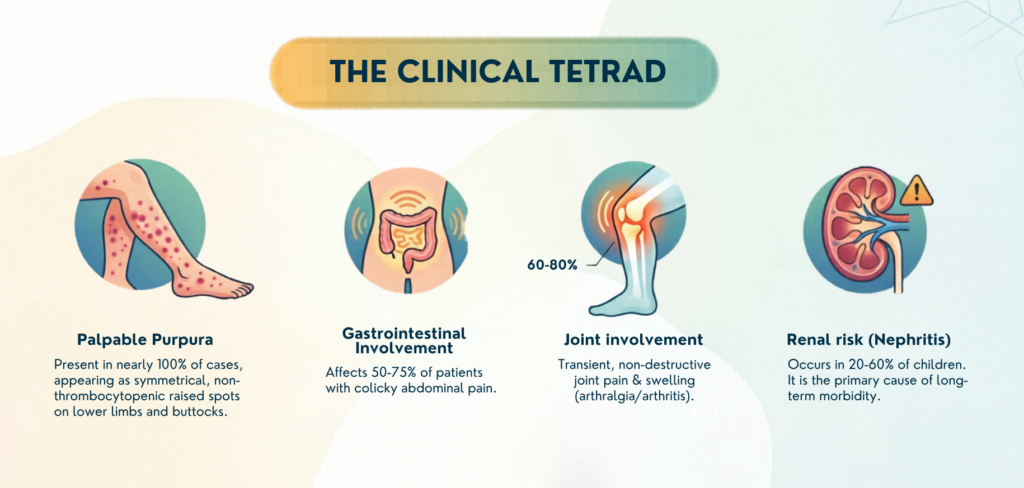
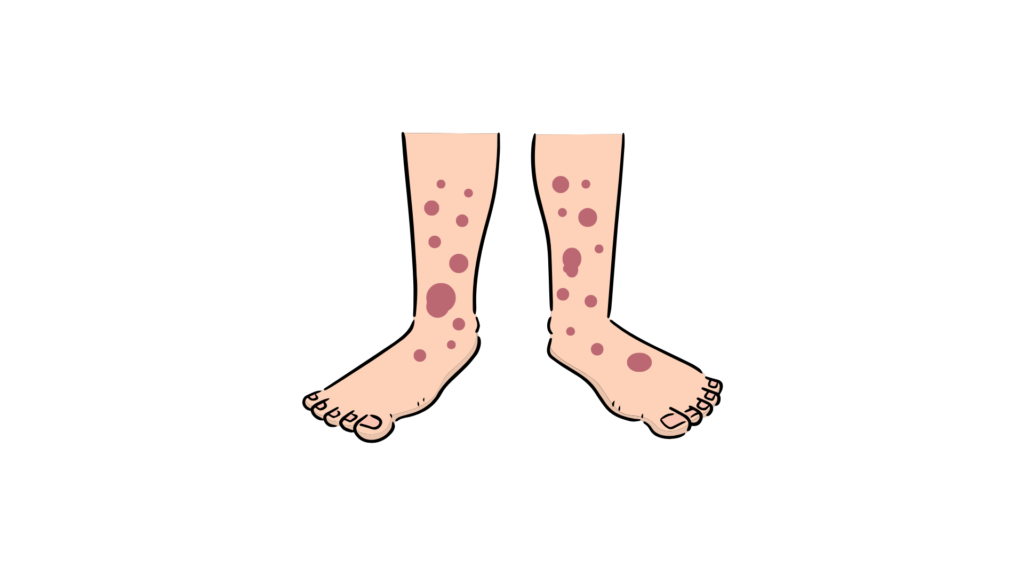
Palpable purpura (Hallmark features)
- Occurring in almost 100% of the patients.
- Typically symmetrical, non-thrombocytopenic petechiae or palpable purpura.
- Predominantly affects the dependent areas – lower extremities (especially extensor surface) & buttocks. Less commonly (atypical) – upper extremities & the trunk.
- Rash often starts as erythematous, macular, or urticarial lesion, later evolving into petechiae/purpura.
- The lesions progress from red –> purple –> rust-colored before fading. These changes occur over approximately 10 days.
- Some patient may develop more severe form of rash i.e. bullous or necrotic, which is associated with higher risk of renal involvement (progressive, treatment-resistant renal failure)
GI involvement
- Symptoms:
– Diffuse (sometimes periumbilical), colicky abdominal pain
– ± nausea, vomiting, diarrhea, GI bleeding (rectal bleeding & melena) - Fecal occult blood test may help detect subclinical GI involvement with no overt bleeding signs.
- Severe complications can include: GI hemorrhage, bowel infarction, perforation or intussusception which require preferential or urgent surgical intervention.
Arthralgia/Arthritis
- Present as painful, swollen joints.
- Typically transient and non-destructive.
- Commonly affects the knees, ankles, feet and hands.
Renal involvement
- The most significant determinant of long-term morbidity & mortality.
- Spectrum ranges from :
a) Microscopic hematuria ± mild proteinuria
b) Nephritic / Nephrotic syndrome
c) Progression to ESRF (in severe cases)
Less common manifestations involving other organs include:
- Neurological : can present with headaches, seizures, mononeuropathy, & in severe cases, intracranial hemorrhage.
- Pulmonary : Rare but potentially fatal pulmonary hemorrhage or interstitial lung disease can occur.
- Others : Orchitis or scrotal edema in boys.
Investigations
There are currently no diagnostic biomarkers for IgA vasculitis.
Diagnosis is primarily clinical, supported by laboratory findings & biopsy when indicated.
Laboratory investigations
- FBC – Assess platelet count to confirm non-thrombocytopenic purpura.
- Renal function tests – Evaluate for kidney involvement/dysfunction.
- Urinalysis with microscopy (Critical investigation) ‼️
- To identify hematuria, proteinuria or RBC casts.
- If urine dipstick is positive for protein, an early morning spot sample should be obtained for albumin-to-creatinine (ACR) or protein-to-creatinine ratios (PCR).
- Others
- Anti-streptolysin O and anti-DNAse B titers -> assess for preceding Group A streptococcal infection.
- Fecal occult blood test – recommended to detect subclinical GI bleed, even without overt digestive bleeding.
Serum IgA are elevated in > 50% of patients but is non-specific with limited diagnostic or prognostic value.
Biopsy & histopathology
a) Skin biopsy
- Not routinely required, but may be considered if the rash is atypical in distribution or severe in nature.
- Typical histopathology : Leukocytoclastic vasculitis with predominant deposition of IgA and complement (C3) within the small vessel walls.
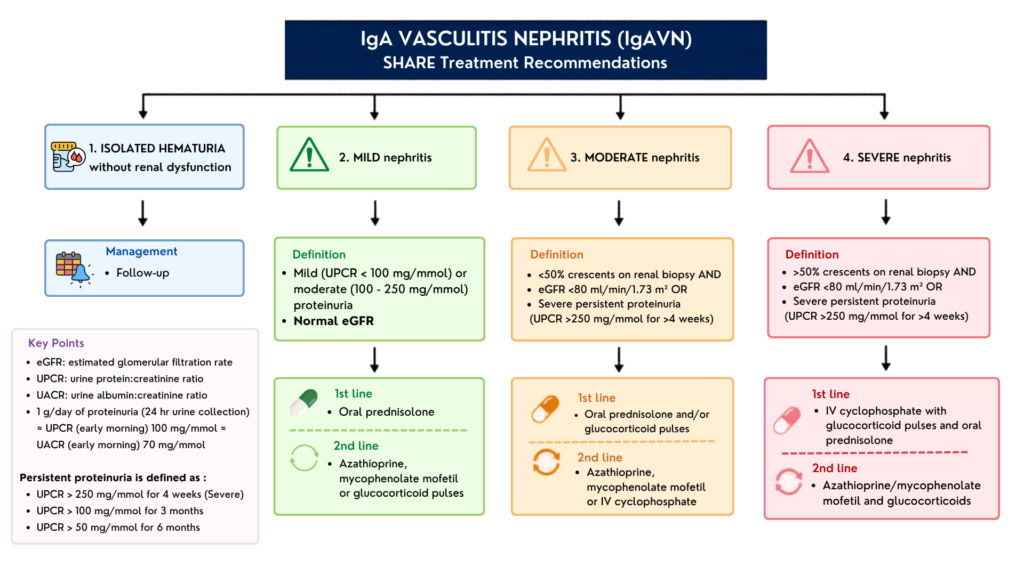
b) Kidney biopsy
Gold standard for definitive diagnosis of IgA vasculitis nephritis.
Absolute indications :
- Severe proteinuria – urine protein-to-creatinine ratio (UPCR) > 250 mg/mmol for at least 4 weeks.
- Persistent moderate proteinuria – uPCR between 100 – 250 mg/mmol for at least 3 months or > 50 mg/mmol for 6 months.
- Impaired eGFR (< 80 mL/min/1.73 m2)
Relative indications :
- Nephritic/nephrotic syndrome
- Clinical signs of rapidly progressive glomerulonephritis (worsening renal function)
- Persistent urine abnormalities – persistent active urinary sediment, or prolonged significant hematuria
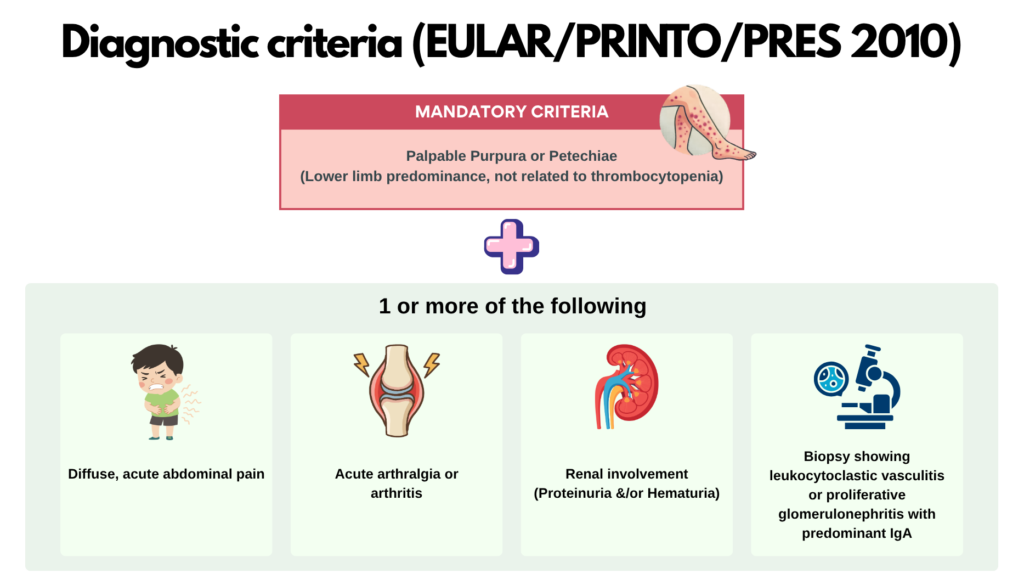
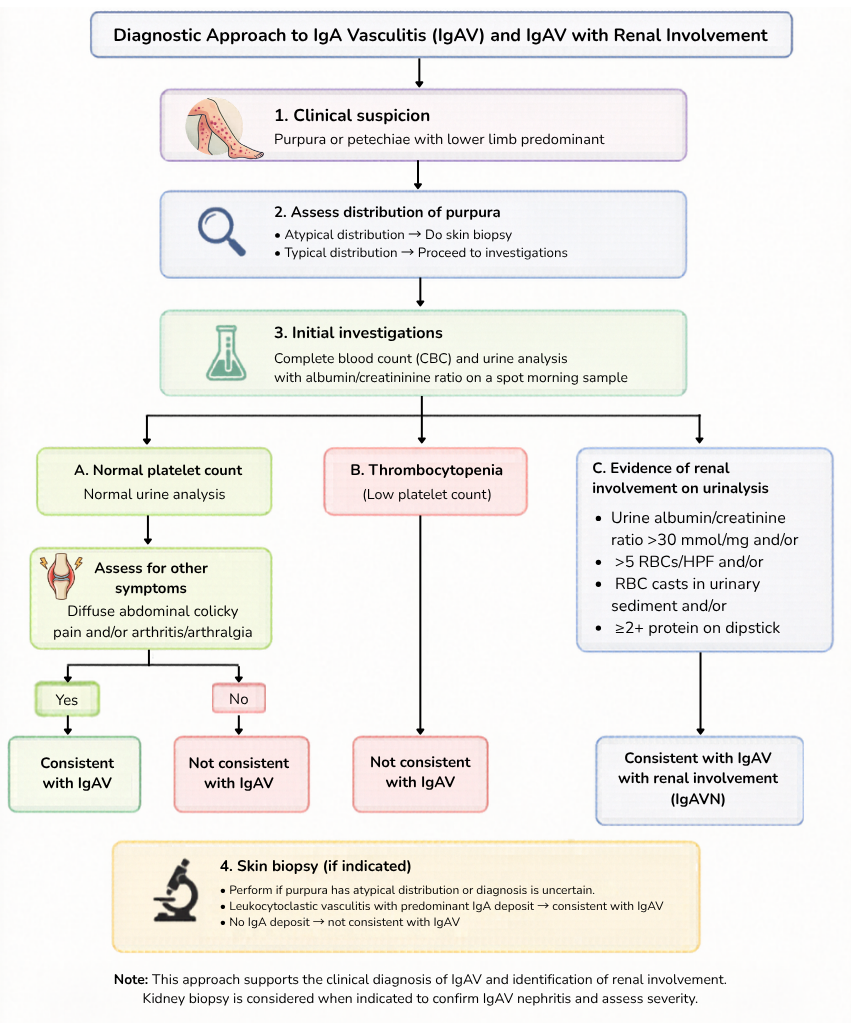
Management
In children, the disease is often self-limiting requiring usually only supportive & symptomatic care, unless renal involvement (& sometimes GI involvement) is present where additional treatment is required. (In contrast to adults which often present with more severe disease requiring aggressive intervention from the start)
1. Supportive & symptomatic care
For mild cases involving the skin & joints, treatment focuses on symptom relief.
General measures : Adequate hydration & rest
Pain management :
- Acetaminophen (PCM) or narcotics are generally preferred over NSAIDs.
- NSAIDs should be avoided if there is active GI or renal involvement due to risk of exacerbating bleeding or worsening renal function.
2. Glucocorticoid therapy
First-line therapy for severe systemic manifestations, including significant GI pain or early-stage renal involvement.
Administration: In children, oral prednisolone is typically used for mild-to-moderate forms, while pulsed IV methylprednisolone is reserved for more severe presentations.
Efficacy: Effective for the rapid resolution of abdominal pain & may reduce the risk of relapses, but do not necessarily prevent the onset of renal disease.
3. Additional treatment (2nd line) – mainly for managing IgAV nephritis
a) Conventional immunosuppressants :
- Mycophenolate Mofetil (MMF) – often used as a steroid-sparing agent in refractory or dependent cases.
- Cyclophosphamide – reserved for severe, rapidly progressive crescentic glomerulonephritis.
- Calcineurin inhibitors (Cyclosporine A or tacrolimus) – may be effective for children with refractory proteinuria.
b) Renin-Angiotensin System (RAS) inhibition (ACEI or ARBs) – critical for those with persistent proteinuria to reduce secondary glomerular damage & manage hypertension.
4. Advanced & Biological Therapies
Consider in life-threatening or refractory situations.
These include :
- Rituximab (RTX) : B-cell depleting agent has shown success in achieving long-term remission in patients who fail conventional therapy.
- Plasmapheresis (PLEX) : Used as an adjunctive therapy for rapidly progressive glomerulonephritis to remove circulating immune complexes.
- Intravenous Immunoglobulin (IVIG) : May be useful in difficult cases, especially when active infection contraindicates traditional immunosuppression.
Prognosis
The prognosis is generally excellent, especially in pediatric populations where the disease is typically self-limiting & usually resolves within 4 weeks.
Long-term morbidity & mortality are almost exclusively determined by the presence & severity of renal involvement.
Short-term prognostic factors
In acute phase of the disease, severe GI manifestations are the leading determinants of short-term prognosis. Life-threatening complications such as intussusception (affecting 3 – 4% of children), bowel perforation, infarction, or massive hemorrhage can be fatal if not managed promptly.
Long-term prognostic factors
Renal involvement, is the only chronic manifestation of the disease & serves as the definitive prognostic factor for long-term health.
Risk & prognosis of renal involvement are dependent on :
a) Age of onset : Adults generally have a poorer long-term prognosis with higher risk of progressing to ESRD.
b) Severity of initial renal presentation (ranging from isolated microscopic hematuria to nephrotic or nephritic syndrome).
c) Predictors associated with increased risk of developing long-term renal complications :
- Skin manifestations : Persistent purpura lasting > 1 month, or necrotic/bullous lesions at onset.
- GI symptoms : Persistent melena or severe GI distress (e.g. intussusception or massive hemorrhage)
- Patient demographics : Adults generally has higher risk of progressing to ESRD.
Follow up
Regular follow up is essential to detect late-onset renal involvement, which can manifest weeks or even months after the initial cutaneous presentation.
Main objective of long-term monitoring is to identify IgA vasculitis nephritis, which remains the most significant chronic complication & the leading cause of morbidity in these patients.
They should be followed up for a minimum of 6 – 12 months, regardless of whether their initial blood pressure (BP) and urinalysis results were normal.
Minimum set of assessment/investigations at each visit :
- BP measurement : to screen for HTN associated with renal involvement.
- Urinalysis with microscopy : to identify microscopic hematuria, proteinuria, or the presence of RBC casts.
- Protein quantification : If urine dipstick is positive for protein, obtain an early morning spot sample to calculate urine protein : creatinine ratio (uPCR) or albumin : creatinine ratio (uACR).
Monitoring schedules (if initial UFEME normal/only microscopic hematuria) :
- Weekly for the 1st month after disease onset
- Every 2 week from week 5 – 12
- Single reviews at 6 and 12 months
- If UFEME remains normal at 12 months, no further follow-up is required.
Indications for referral to Pediatric Nephrologist
- Gross hematuria
- Acute nephritis
- Abnormal proteinuria with or without nephrotic syndrome (including sub-nephrotic range)
- Presence of hypertension
- High serum creatinine
References
- Roache-Robinson P, Killeen RB, Hotwagner DT. IgA Vasculitis (Henoch-Schönlein Purpura) [Updated 2023 Sep 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537252/
- Castañeda, S., Quiroga-Colina, P., Floranes, P., Uriarte-Ecenarro, M., Valero-Martínez, C., Vicente-Rabaneda, E. F., & González-Gay, M. A. (2024). IgA Vasculitis (Henoch-Schönlein Purpura): An Update on Treatment. Journal of clinical medicine, 13(21), 6621. https://doi.org/10.3390/jcm13216621
- Sestan, M., & Jelusic, M. (2023). Diagnostic and Management Strategies of IgA Vasculitis Nephritis/Henoch-Schönlein Purpura Nephritis in Pediatric Patients: Current Perspectives. Pediatric health, medicine and therapeutics, 14, 89–98. https://doi.org/10.2147/PHMT.S379862
- Ministry of Health Malaysia, & Malaysian Paediatric Association. (2025). Paediatric protocols for Malaysian hospitals (5th ed.). Ministry of Health Malaysia.